

Separating the molecular mechanisms that regulate the onset of ductal plate formation from those controlling differentiation of hepatoblasts to cholangiocytes remains difficult. The role of TGFβ, Notch, Wnt, Fgf, Laminin, Hippo and Epimorphin signalling in cholangiocyte differentiation has been reviewed elsewhere,22, 80, 81, 82 but how these pathways control subsequent tubular network formation has yet to be discussed. TGFβ receptor II and TGFβ signalling are activated in hepatoblasts differentiating to ductal plate cholangiocytes, but they become repressed once the cholangiocytes delineate a lumen. Such repression is supported by direct and indirect observations. Indeed, inhibition of TGFβ represses formation of biliary-like cysts in an in vitro morphogenesis assay,83 while persisting receptor expression is associated with delayed duct maturation in SRY-related HMG box transcription factor (Sox)4- and Sox9-deficient livers.73, 84
Blocking Notch signalling by timed gene inactivation or pharmacological inhibition demonstrated that Jagged 1 and Notch 2, whose mutations cause bile duct paucity in patients affected by Alagille syndrome, are required for tube formation.52, 85, 86, 87 Notch signalling initially induces differentiation of the single-layered ductal plate from hepatoblasts, following an interaction between the Jagged 1-positive portal mesenchyme and Notch 2 in hepatoblasts.87 Notch then promotes tubulogenesis, most likely through Jagged-Notch interactions occurring between the cholangiocytes on the portal side and the hepatoblast-like cells located on the parenchymal side of the early luminal structures.85 Therefore, in parallel to growth along the longitudinal axis of the ducts, differentiation proceeds radially. Genetic modifiers of Notch signalling are suspected to modulate the disease phenotype, but their identity remains elusive. However, a possible candidate is o-glycosyltransferase 1 (Poglut1, also called Rumi), which was recently discovered to post-translationally block Jagged 1 function.88 POGLUT1 may thereby act as genetic modifier of Alagille syndrome, since its inactivation partially compensates for the duct paucity induced by heterozygous Jagged 1 mutations. Other potential genetic modifiers are Hnf6 and the fringe family of glycosyltransferases.89, 90
Ductal plate-specific inactivation of β-catenin revealed that canonical Wnt signalling is dispensable for normal duct formation. Yet, the level of signalling must be tightly controlled, because β-catenin overexpression strongly perturbs ductal morphogenesis.91 Similarly, non-canonical Wnt5a signalling needs to be fine-tuned since it represses cholangiocyte differentiation in vivo and reduces tube size in in vitro experiments.92
Finally, studying ECM-cholangiocyte interactions highlighted the differentiation- and morphogenesis-promoting functions of collagens and of several laminins in vitro.83, 93, 94, 95, 96, 97 However, less is known from in vivo functional studies, except that Integrinβ1 is required throughout differentiation and morphogenesis, and is activated by Laminin α5.98 Laminin α5 is necessary for biliary morphogenesis and is produced by the ducts, indicating that ducts activate a genetic programme sustaining their own morphogenesis.
Cholangiocyte polarity and duct morphogenesis
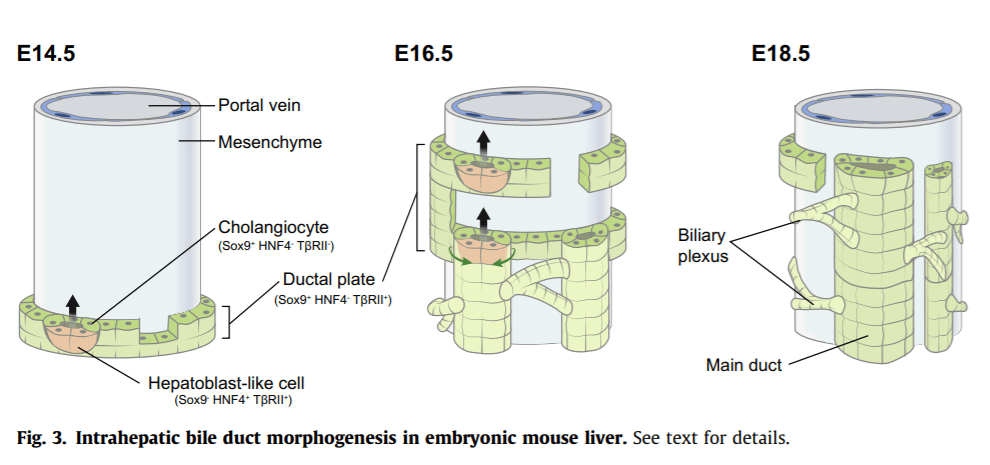
Polarisation is a hallmark of maturing cholangiocytes.83 The earliest ductal plate cells are irregularly shaped but become cuboidal as soon as they line luminal structures. Apicobasal polarity then becomes evident as the primary cilia develop and Spp1 is expressed at the apical side; tight junctions separate the apicolateral and basolateral domains, the latter marked by high expression of E-cadherin. At the initial stage, i.e. when duct lumina are asymmetrically delineated by cholangiocytes and hepatoblast-like cells, the latter exhibit low E-cadherin levels while already expressing Spp1. This indicates that apicobasal polarisation is stimulated as soon as lumina are generated, even in cells lining the lumina which do not yet express typical cholangiocyte markers such as Sox9.73 Some apical proteins such as ezrin display biliary-specific expression in developing livers and are not found in hepatocytes.99
The morphogenic importance of tight junctions is underscored by the role of claudins, which are core constituents of tight junctions and whose mutations in humans cause sclerosing cholangitis.100 Analyses in developing zebrafish revealed that mutations in claudin 15-like b (cldn15lb) perturb bile duct remodelling, hepatocyte polarisation and bile canaliculi formation, suggesting that these three processes are tightly linked.46, 64 This potential link is further supported by the observation that cholestasis and abnormal duct development are caused by mutations in VPS33B, VIPAR, Vps18 and Atp6ap2, four genes coding for proteins controlling intracellular trafficking and hepatocyte polarisation.61, 62, 63, 101, 102, 103 Similarly, blocking canaliculi formation by inhibition of Mrp2 prevents remodelling of the ducts into a mature biliary plexus.52 Along the same lines, duct tubulogenesis is inhibited in mice deficient in Lkb1, which is critical for normal canalicular structure and function.60, 104 Considering that initiation of bile flow through developing ducts coincides with duct remodelling, it is suggested that the influx of bile constitutes the actual trigger for bile duct remodelling.52
Beyond tube formation and initiation of bile transport, epithelial maturation of cholangiocytes proceeds by strengthening the barrier function of tight junctions. This is controlled by the transcription factor Grainyhead-like 2, which stimulates expression of claudins and Rab25, with the latter promoting the localisation of Claudin 4 at the tight junction.105, 106
E-cadherin expression is strongly enhanced at the basolateral side starting at the onset of cholangiocyte differentiation. Still, E-cadherin is required for adult biliary homeostasis, but not for morphogenesis.107 Despite p120-catenin being well known for its E-cadherin-stabilising properties, it is dispensable for E-cadherin expression in cholangiocytes. Yet, its absence causes development of cholangiocytes displaying a hybrid hepatocyte-cholangiocyte phenotype, resulting in ductal plate malformation and lack of normal duct development.108
Planar cell polarity (PCP), which regulates directional cell behaviour, also controls tubulogenesis. The role of PCP in bile duct morphogenesis is supported by the observation that inhibition of genes typically involved in PCP, namely prickle-1a, vangl2 and ankrd6, reduces the number of ducts and duct interconnections in developing zebrafish liver.109 However, this is associated with defective vesicle transport in hepatocytes and abnormal bile formation, suggesting that perturbed bile flow may, again, contribute or even cause abnormal duct morphogenesis in these morphant livers. Still, the common bile duct, which belongs to the extrahepatic duct system, is shortened and shows fewer mucosal folds in mice with Prickle mutations, thereby extending the role of prickle to mammalian biliary tree development.110 Interestingly, this study revealed that a prickle mutation also causes defective development of primary cilia, which are connected both with PCP and apicobasal polarity.110 Primary cilia are at the origin of a wide range of diseases best known as ciliopathies, a number of which affect biliary development and cause aberrant cholangiocyte differentiation, ductal plate malformations, absence of bile ducts, and polycystic or dysplastic bile ducts.76, 80, 111
Bile duct morphogenesis in response to injury
Cholestatic injury forces the liver to cope with bile, the accumulation of which may eventually become toxic. Following bile duct ligation, the cholangiocytes lining terminal branches of interlobular ducts proliferate. Elongation and branching of ducts is stimulated, parallelled by a wrinkling of the epithelium (corrugation) leading to an increase in luminal surface. When injury persists over time, elongation and corrugation are amplified, and branched structures establish multiple connections; large bile ducts increase their diameter, unlike terminal ducts whose diameter remains unaffected.112 Collectively, these morphological responses optimise bile absorption in the terminal branches and fluid drainage in larger ducts.
The cholestasis-induced morphological response differs from toxin-mediated injury of the liver. Indeed, Itoh and coworkers studied bile duct remodelling triggered by a variety of hepatotoxic agents, extending earlier findings on ANIT-treated rats and elegantly illustrating, at high resolution, how bile duct anatomy adapts to the nature of the toxic injury in mice.75, 113 Periportal hepatocyte damage induces a major expansion of the periportal biliary plexus, whereas pericentral hepatocyte death causes linear development of biliary branches towards the necrotic area. A combination of three-dimensional analysis, single-cell tracing and mathematical simulations has shown that cholangiocytes are heterogenous in terms of proliferative capacity: cholangiocytes lining peripheral ductules proliferate more upon injury than those lining larger ducts. Moreover, not all peripheral cholangiocytes proliferate and those that do generate clones of variable size, indicating that biliary remodelling results from a stochastic mode of response to injury rather than from a fixed pattern of stem cell-dependent tissue response.114 The benefit for the liver of such biliary remodelling is not clear. However, the expanding ducts and plexus may constitute a source of growth factors for hepatocyte regeneration, or provide a frame for re-establishing contacts between hepatocytes and cholangiocytes.75, 115 The biliary response is expected to vary according to the nature of the hepatic injury, since, depending on the severity of hepatocyte damage biliary cells will or will not transdifferentiate towards hepatocytes to restore liver function, as shown in zebrafish and more recently in mice.116, 117, 118, 119
Development of the extrahepatic biliary tree
The anatomy of the extrahepatic biliary tract, which comprises the gallbladder and the common, cystic and hepatic ducts, is similar in mice and humans,120 but its ontogeny did not attract as much attention as intrahepatic duct development. However, it is now well demonstrated that the extrahepatic ducts do not derive from the liver. Instead, they have a common origin with the ventral pancreas and develop as an elongated outgrowth of the ventral endoderm.121 Tan and Moscoso showed that the extrahepatic ducts are located in the vicinity of the intrahepatic biliary ducts at the hilum of the developing human liver. Yet, how the intra- and extrahepatic ducts anastomose is still not clear.122 The most distal parts of the extrahepatic ducts are likely to introgress slightly into the hilum of the liver and the largest hilar ducts of an adult liver may actually arise from the extrahepatic biliary tree. This is supported by our observation that inactivation of Sox4 and Sox9 in hepatoblasts preserves the hilar ducts while abolishing hepatoblast-derived peripheral duct formation.84 Concordantly, the mutational spectrum associated with tumourigenesis is similar in hilar and extrahepatic cholangiocarcinoma, but different to that of intrahepatic cholangiocarcinoma, suggesting that the hilar and extrahepatic duct have a common gene expression background. This is expected to confer similar susceptibility to cancer and likely results from the hilar and extrahepatic ducts having a common embryonic origin.123
Investigations into transcriptional regulation in extrahepatic ducts revealed that they originate from progenitors co-expressing the transcription factors Pdx1 and Sox17.121, 124 Sox17 is required to separate the prehepatic endodermal domain from the prepancreatic. In the absence of Sox17, the extrahepatic biliary tree fails to develop and is replaced by pancreatic tissue. Haploinsufficient Sox17 mice show hypoplastic gallbladder epithelium, and conversely, overexpression of Sox17 promotes ductal development.125 Sox17 is involved in a feedback loop with hairy and enhancer of split 1 (Hes1), another factor whose absence causes gallbladder agenesis.126, 127 Sox17 is epistatic to Hhex, Hnf1β, Hnf6, and likely also to Forkhead box factor 1 (Foxf1) and Leucine-rich repeat-containing G-protein coupled receptor 4 (Lgr4), all genes whose loss-of-function causes various forms of extrahepatic biliary dysmorphogenesis, such as gallbladder agenesis, common bile duct enlargement, epithelial dysplasia or replacement by duodenal tissue.128, 129, 130, 131, 132
Congenital diseases of the extrahepatic ducts are rare and their aetiology is in most cases unknown. Yet their investigation is likely to uncover new mechanisms of extrahepatic duct development. Nf2 mutations in zebrafish are associated with choledochal cyst formation. In light of the repression exerted by Nf2 on the hippo effector Yap in intrahepatic ducts,103, 133 it is tempting to speculate that an Nf2-Hippo-Yap cascade modulates common bile duct growth. Clinically, the most important extrahepatic duct malformation is biliary atresia, a syndrome whose aetiology is still debated and for which several susceptibility genes have been proposed.24 An in-depth description of this syndrome is out of the scope of the present review. However, its study revealed the role of Glypican 1 (Gpc1), a heparan sulfate proteoglycan that potentially binds FGF19 and Hedgehog ligands. GPC1 is indeed a biliary atresia susceptibility gene and its knock down in zebrafish results in intrahepatic duct paucity and gallbladder hypoplasia.134 Finally, the identification of biliatresone as a toxin that induces biliary atresia has revealed the importance of well-controlled redox homeostasis for normal extrahepatic bile duct development.135
Origin of hepatic endothelial cells
The morphogenetic steps through which the hepatic venous system is patterned in human and mouse embryos have been extensively described.136, 137, 138, 139 According to the “vestigial” theory, the umbilical and vitelline veins give rise to the hepatic venous system. However, this theory was questioned in humans since the vitelline veins are not functional and only the left umbilical vein is present at the stage of liver organogenesis.136 This suggests that the venous system is essentially shaped by the blood flow from the left umbilical vein.
Several sources of sinusoidal endothelial cells have been considered. Lineage tracing in mouse embryos did not provide evidence for septum transversum-derived vasculature.38 However, in the avian embryo, there is evidence that mesothelial cells invade the liver and contribute in part to liver sinusoidal endothelial cells (LSEC).140 More recent studies in mice have found that the endocardium of the sinus venosus, which is located near the liver diverticulum and hepatic bud at E 8.0-8.5, contributes no less than a third of the endothelium lining the sinusoids, the portal and the central veins.141 The sinus venosus flanks the liver at the budding stage. The sinus venosus-derived cells are attracted to the early liver by Vegf secreted by hepatoblasts, and they persist into adulthood where they participate in liver regeneration following injury.13, 141 In parallel, lineage tracing using Foxa2:T2AiCre or Foxa2:CreER mice, which enables labelling of endoderm-derived cells, suggests that the endoderm also contains progenitors for a subset of endothelial cells.142 The percentage of endoderm-derived endothelial cells was estimated to be around 15%, using Foxa2:T2AiCre mice. This number must be considered with caution since Foxa2:T2AiCre also labels cardiac cells located close to the developing liver,143 and whether Foxa2:T2AiCre-labelled cells overlap with sinus venosus cells has not been verified. Moreover, when human or mouse embryonic stem cells are programmed in vitro towards an endoderm fate and subsequently grow in hepatic-inducing medium, a population of endothelial cells emerges with residual Hnf4 expression, a marker of endoderm-derived hepatoblasts. Similarly, sections of human foetal liver specimens also show cells double positive for HNF4 and endothelial marker CD31.144 Together, these data indicate that part of the liver endothelial cells are derived from the sinus venosus and endoderm, and possibly mesothelium. Vitelline veins are located in the vicinity of the liver and share a common marker with sinusoidal cells (Stabilin 2), suggesting that they can also contribute to liver endothelial cell development.145, 146 In zebrafish, elegant lineage tracing studies demonstrated that the liver vasculature is essentially of venous origin.147
Morphogenesis of veins, arteries and sinusoids
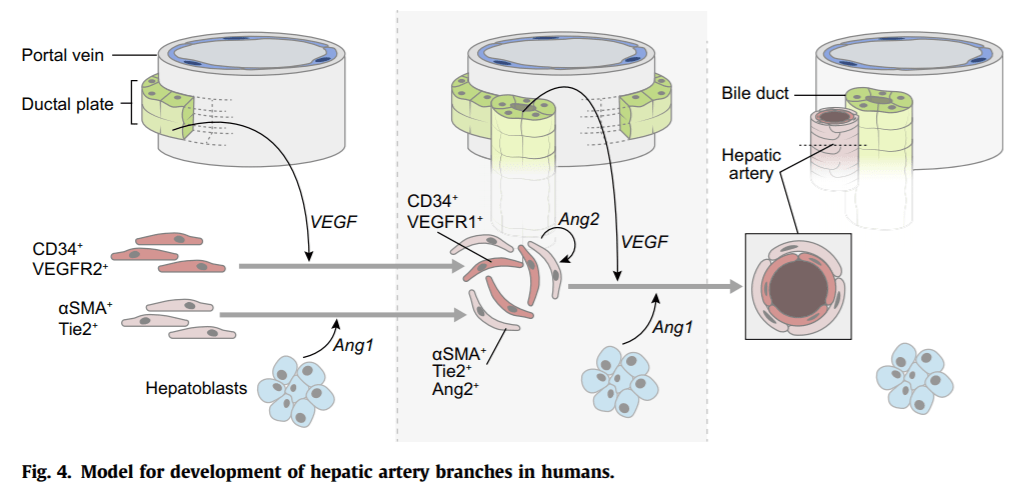
The spatial organisation of blood flow in adult liver lobules is well known and recent improvements in casting, imaging and modelling provide additional quantitative information regarding the three-dimensional architecture of the liver vasculature.148, 149, 150 However, it remains unclear how the branching pattern is established during embryogenesis and whether vessel morphogenesis results from angiogenesis or vasculogenesis. The portal vein is the first to organise a branched structure, constituting a frame for development of the biliary tree, hepatic artery branching and haematopoietic stem cell expansion. The portal mesenchyme instructs ductal plate development (see earlier), and biliary cells then promote hepatic artery morphogenesis.151, 152 Expression data suggest that artery development depends on VEGF and Angiopoietin according to a sequence of events summarised (Fig. 4). VEGF-A is expressed widely in developing liver and most strongly in ductal plate cells.152, 153 It recruits VEGFR-2-positive endothelial cells to the portal mesenchyme near the ductal plate, where they aggregate as VEGFR-1-positive endothelial cells. The latter assemble into vascular structures with myofibroblast-derived mural cells expressing TIE-2, a receptor for Angiopoietin-1 secreted by hepatoblasts. Arterial remodelling is then likely promoted by autocrine secretion of Angiopoietin-2. In parallel, a vascular peribiliary plexus develops, but its origin remains debated as it may result from branching of the hepatic artery or from local vasculogenesis.153, 154 In line with this model, inactivation of VEGF-A in mid-gestational hepatocytes and cholangiocytes causes a reduction in the number of hepatic endothelial cells.155 The interaction between developing bile ducts and hepatic arteries may differ slightly in humans and mice, since arterial morphogenesis in humans occurs along the ductal plate, whereas in mice, arteries only form postnatally and alongside mature bile ducts.151

{kind=link}
{kind=link}
{kind=link}