
Growth Hormone Stimulates Proliferation of Old-Aged Regenerating Liver Through Forkhead Box m1b
From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways
20.09.2019
Improvement of the Modeling and Diagnosis of Hepatic Encephalopathy in Rats
20.09.2019
Authors: Katherine Krupczak-Hollis, Xinhe Wang, Margaret B. Dennewitz, and Robert H. Costa.
Abstract
The Forkhead Box (Fox) proteins are an extensive family of transcription factors that shares homology in the winged helix DNA-binding domain and the members of which play essential roles in cellular proliferation, differentiation, and longevity. Reduced cellular proliferation during aging is associated with a progressive decline in both growth hormone (GH) secretion and Foxm1b expression. Liver regeneration studies with 12-month-old (old-aged) transgenic mice indicated that increased hepatocyte expression of Foxm1b alone is sufficient to restore hepatocyte proliferation to levels found in 2-month-old (young) regenerating liver. GH therapy in older people has been shown to cause an increase in cellular proliferation, but the transcription factors that mediated this stimulation in proliferation remain uncharacterized. In this study, we showed that human GH administration to old-aged Balb/c mice dramatically increased both expression of Foxm1b and regenerating hepatocyte proliferation. This increase in old-aged regenerating hepatocyte proliferation was associated with elevated protein expression of Cdc25A, Cdc25B, and cyclin B1, with reduced protein levels of cyclin-dependent kinase inhibitor p27Kip1 (p27). GH treatment also was found to stimulate hepatocyte proliferation and expression of Foxm1b protein without partial hepatectomy (PHx). Furthermore, GH treatment of young Foxm1b -/- mice failed to restore regenerating hepatocyte DNA replication and mitosis caused by Foxm1b deficiency. These genetic studies provided strong evidence that the presence of Foxm1b is essential for GH to stimulate regenerating hepatocyte proliferation. In conclusion, our old-aged liver regeneration studies show that increased Foxm1b levels are essential for GH to stimulate hepatocyte proliferation, thus providing a mechanism for GH action in the elderly. (HEPATOLOGY 2003; 38:1552-1562.)
Cell division is tightly regulated, especially at the G1/S-phase (DNA replication) and G2/M-phase (mitosis) transitions, by temporal activation of multiple cyclin-dependent kinases (Cdk) complexed with their corresponding cyclin regulatory subunits. In addition to assembly with a cyclin regulatory subunit, Cdk activity requires dephosphorylation of the Cdk catalytic subunit by the Cdc25A, Cdc25B, or Cdc25C phosphatases.1-3 Furthermore, Cdk inhibitors from the CIP/KIP family (p21Cip1 and p27Kip1 [p27]), and the INK4 family (16INK4A) become elevated during aging4-8 and negatively regulate Cdk activity. Progression into S-phase requires activation of Cdk2 in complex with either cyclin E or cyclin A that cooperates with cyclin D-Cdk4/6 to phosphorylate the Retinoblastoma (RB) protein, which releases bound E2F transcription factor and allows it to stimulate expression of genes required for DNA replication.9,10 At the end of S-phase, activation of Cdk1 requires the removal of inhibitory phosphates at Thr 14 and Tyr 15 by the Cdc25B phosphatase and decreased phosphorylation of these residues by the Myt1/Wee1 kinases.1,11,12 Likewise, phosphorylation of critical target proteins by the active cyclin B-Cdk1 complex mediates progression into mitosis.13
The Forkhead Box (Fox) transcription factors are an extensive family of transcription factors, consisting of more than 50 mammalian proteins,14 that shares homology in the winged helix DNA-binding domain.15 Its members play important roles in regulating expression of genes involved in cellular proliferation,16,17 differentiation,18,19 apoptosis,20 transformation,21 longevity,22 and metabolic homeostasis.23 The human Foxm1b transcription factor (previously known as HFH-11B, Trident, Win, or MPP2) is expressed in proliferating cells, but its expression is extinguished when the cells undergo terminal differentiation and exit the cell cycle.24-27
The mammalian liver is one of the few adult organs capable of completely regenerating itself in response to injury without using a stem cell population in which growth factors and cytokines stimulate reentry of terminally differentiated hepatocytes into the cell cycle.28-31 In regenerating liver, increased hepatocyte expression of Foxm1b occurs at the G1/S transition of the cell cycle and its levels remain elevated throughout the period of proliferation.27 Premature hepatocyte expression of FoxM1B in regenerating liver of transgenic or FoxM1B adenovirus– infected mice accelerated the onset of hepatocyte DNA replication and mitosis by stimulating earlier expression of cell cycle genes.16,32 Concurrent with this, increased FoxM1B expression in regenerating livers of 12-monthold (old-aged) mice is sufficient to restore hepatocyte proliferation to levels found in young regenerating mouse liver, reduce levels of Cdk Inhibitors p27 protein, and increase expression of M-phase promoting cyclin B and Cdc25B.33,34 Furthermore, we used the albumin promoter/enhancer–driven Cre recombinase transgene (Alb-Cre) to mediate hepatocyte-specific deletion of the Foxm1b Floxed (fl) allele, and Foxm1b deficiency caused significant reduction in regenerating hepatocyte proliferation.17 Diminished proliferation of young regenerating Alb-Cre Foxm1b -/- hepatocytes was associated with increased protein levels of Cdk inhibitor p21Cip1 and reduced protein expression of Cdc25A and Cdc25B phosphatase, leading to decreased Cdk2 and Cdk1 activation.17 These studies indicated that FoxM1B controls cell cycle progression at both the S-G1 and G2-M phase transitions during liver regeneration and that increased expression of FoxM1B in old-aged mice is sufficient to reestablish proliferation of regenerating hepatocytes.
During aging, reduced cellular proliferation leads to diminished muscle and bone mass, defective tissue repair, and thinning of the skin, which is associated with a progressive decline in growth hormone (GH) secretion35-37 and FoxM1B expression.33,38 GH therapy in the elderly is known to stimulate cellular proliferation and was accompanied by a statistically significant increase in muscle (lean body) mass, skin thickness, and average lumbar vertebral bone density while causing decrease in adipose tissue mass.37 Furthermore, GH treatment of young rats has been shown to stimulate hepatocyte DNA replication and mitosis after partial hepatectomy (PHx),39 but the transcription factors required for GH-mediated stimulation in hepatocyte proliferation remain uncharacterized. In this study, we showed that administration of human GH to 12-month-old (old-aged) Balb/c mice restored regenerating hepatocyte proliferation and Foxm1b expression to levels similar to those found in young regenerating liver. This stimulation of old-aged regenerating hepatocyte proliferation was associated with increased expression of Cdc25A and Cdc25B phosphatases and cyclin B1 protein with significant reduction in p27 protein levels, several of which were changes previously attributed to restoring FoxM1B levels.33,34 Consistent with the role of Foxm1b in mediating this increase in proliferation, GH treatment of young Alb-Cre Foxm1b -/- mice failed to stimulate the reduction in regenerating hepatocyte DNA replication and mitosis caused by Foxm1b deficiency. These studies showed that restoring GH levels in old-aged mice mediated an increase in regenerating hepatocyte proliferation by elevating expression of Foxm1b and, thus, provide a mechanism for GH action in the elderly.
Materials and Methods
PHx, Immunohistochemical Staining, and Human GH Treatment. Untreated 2-month-old (young) and 12-month-old (old-aged) Balb-c mice (purchased from the National Institute on Aging) or human GH-treated old-aged Balb-c mice were subjected to PHx to induce liver regeneration.16,33 One hour before PHx, old-aged Balb-c mice were given intraperitoneal (IP) injections of 4 µg/g body weight of human GH resuspended in phosphate-buffered saline (Somatropin [Norditropin]; Novo Nordisk Pharmaceuticals Inc., Princeton, NJ). Subsequent IP injections of human GH were administered every 8 hours (at 3 µg/g body weight) for the duration of liver regeneration. An IP injection of phosphate-buffered saline containing 10 mg/mL 5-bromo-2`-deoxyuridine (BrdU) (Sigma; 50 µg/g body weight) was administered to all mice 2 hours before harvesting the remnant regenerating liver. Three mice at each timepoint were sacrificed using CO2 asphyxiation at the following intervals after PHx: 24, 32, 36, 40, 44, and 48 hours. The regenerating livers were harvested and divided into 3 portions to isolate total RNA,16 to isolate total protein extract, and for paraffin embedding.40 Hepatocyte DNA replication was monitored by immunohistochemical detection of BrdU incorporation (Roche) of 5 µm regenerating liver sections with a monoclonal antibody against BrdU. The mean number of BrdU-positive cells per 1,000 hepatocytes (SD) was calculated by counting BrdU-positive hepatocytes from 3 regenerating mouse livers as described previously.16,33 To determine the number of hepatocytes undergoing mitosis, regenerating liver sections from 3 mice at 36, 40, 44, and 48 hours after PHx were stained with hematoxylin and eosin and the number of mitotic figures per 1,000 hepatocytes SD was calculated as described previously.16,33 Antibodies specific to FoxM1B,16,27Cdc25B, or cyclin B1 (Santa Cruz Biotechnology, Santa Cruz, CA) proteins were used for immunohistochemical detection of 5 µm regenerating liver sections using methods described previously.16,27,34 Twomonth-old and 12-month old Balb/c mice were given initial IP injections of 4 µg/g body weight of human GH (3 mice of each were used). Subsequent IP injections of human GH were given every 8 hours (at 3 µg/g body weight) until they were sacrificed at 41 hours after the initial GH injection. BrdU was administered 2 hours before sacrificing the GH-treated mice, and liver sections were prepared and used for immunohistochemical staining to measure BrdU incorporation and determine levels of hepatocyte DNA replication as described previously in this article. National Institutes of Health guidelines for animal care were followed.
Western Blot Analysis and Ribonuclease (RNase) Protection Assays. For Western blot analysis, 50 µg of total protein extracts prepared from regenerating liver40 were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Protran membrane (Schleicher & Schuell, Keene, NH). The signals from primary rabbit antibodies specific to either p27 (Cell Signaling, Berkeley, CA), Cdc25A (Santa Cruz Biotech, 1:200), or Cdc25B (Santa Cruz Biotech, 1:500) proteins and mouse antibodies specific to either FoxM1B (1:5,000) or -actin (clone AC-15; Sigma; 1:30,000 dilution) proteins were amplified by biotin-conjugated anti-rabbit or anti-mouse immunoglobulin G (Bio-Rad, Hercules, CA), and detected with Enhanced Chemiluminescence Plus (Amersham Pharmacia Biotech, Piscataway, NJ). To produce the mouse antibody against human FoxM1B protein, human FoxM1B C-terminal region (amino acids 365 to 748) was cloned into the p28A expression vector (Novagen) in frame with the Histidine (His)-tagged epitope. The His-tagged FoxM1B 365-748 protein was expressed in BL21 DE3 bacteria, protein extracts were made, and the His-tagged FoxM1B 365-748 protein was affinity-purified on Nickel column (Novagen) using the manufacturer’s protocol. Mice were immunized with the affinity purified His-tagged human FoxM1B 365-748 protein, and mouse serum was isolated by the University of Illinois at Urbana/Champagne Immunological Resource Center.
Total RNA was prepared from mouse liver at the indicated hours after PHx with RNA-STAT-60 (Tel-Test B Inc., Friendswood, TX) and was used for RNase protection assays with antisense (-32P) UTP-labeled probes specific to either the Foxm1b or Cdc25B genes. Expression levels of these genes were calculated as described previously.17,32-34
Generation of Mice With fl Foxm1b–Targeted Allele, Hepatocyte-Specific Deletion, and GH Treatment After Phx. Generation of the mice containing the Foxm1b LoxP/LoxP (fl/fl)-targeted allele, which were bred into the C57/B6 background for 4 generations, has been described previously.17 Conditional deletion of the Foxm1b fl/fl allele in adult hepatocytes was accomplished through breeding with the Alb-Cre C57B6 transgenic mice41 (Jackson Labs). Eight-week-old (young) Foxm1b fl/fl or Alb-Cre Foxm1b -/- mice were subjected to PHx, and 3 mice were killed at 40, 44, and 48 hours after surgery by CO2 asphyxiation and examined for BrdU incorporation and mitosis as described previously in this article. Young Alb-Cre Foxm1b -/- mice also were administered human GH before PHx and treated with human GH every 8 hours until the regenerated liver was harvested as described previously in this article.
Results
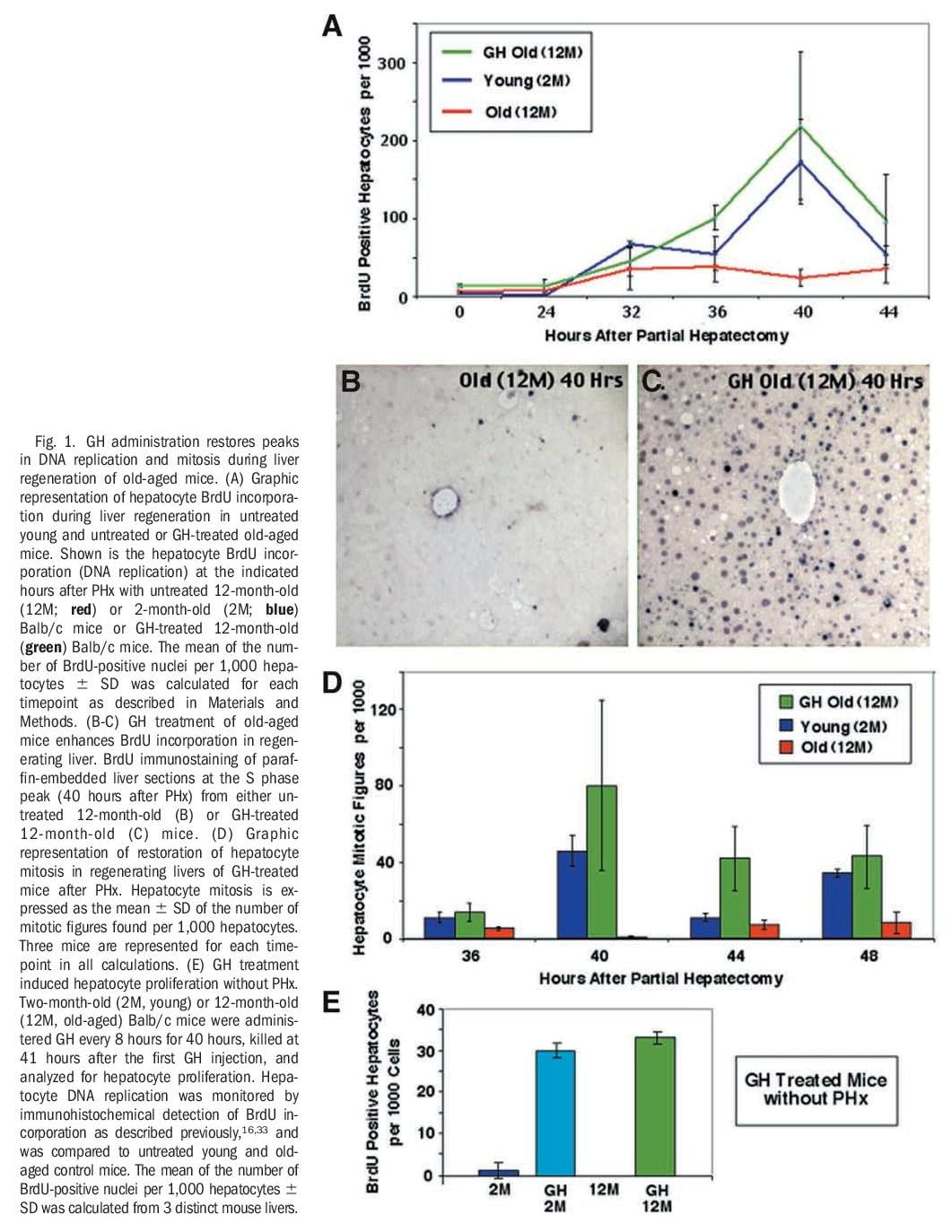
Human GH Treatment of Old-Aged Mice Increases Regenerating Hepatocyte Proliferation. Reduced cellular proliferation during aging is associated with a progressive decline in GH secretion35-37 and FoxM1B expression.33,38 Restoring hepatocyte expression of FoxM1B alone in 12-month-old (old-aged) mice is sufficient to increase regenerating hepatocyte proliferation and expression of cell cycle regulatory genes to levels found in young (2-month-old) regenerating mouse liver.33 Therefore, we wanted to examine whether administering human GH to old-aged mice would increase regenerating hepatocyte proliferation and expression of Foxm1b. To test this hypothesis, we compared regenerating hepatocyte proliferation in untreated young and old-aged Balb/c mice with that of human GH–treated old-aged mice at various timepoints after PHx as described in Materials and Methods. Hepatocyte DNA synthesis was monitored by immunohistochemical staining for BrdU incorporation, and hepatocyte mitotic figures were measured in hematoxylin and eosin–stained regenerating liver sections.16,33 Young regenerating livers displayed initiation of hepatocyte DNA replication at 32 hours after PHx with a BrdU incorporation peak at 40
hours after PHx (Fig. 1A, blue line) and they exhibited mitosis between 40 to 48 hours after PHx (Fig. 1D, blue bar). As reported previously,34 untreated old-aged regenerating livers displayed a significant reduction in hepatocyte DNA synthesis (Fig 1A, red line) and mitosis (Fig 1D, red bar) compared with young regenerating liver. In contrast, human GH treatment of old-aged mice increased hepatocyte BrdU incorporation (DNA replication, Fig. 1B-C) and mitosis to levels found in young regenerating liver (Fig. 1A and D, compare green and blue).
Next, we determined whether GH administration to either young (2-month-old) or old-aged (12-month-old) Balb/c mice is sufficient to induce hepatocyte proliferation in the absence of liver injury. Young and old-aged Balb/c mice were treated with GH every 8 hours without PHx and, then, livers were harvested at 41 hours after the initial GH injection. Two hours before sacrificing the GH-treated mice, we injected them with BrdU to determine levels of hepatocyte DNA replication by measuring BrdU incorporation. These studies showed that GH injections were sufficient to induce hepatocyte DNA replication in both young and old-aged mice without PHx (Fig. 1E), but at lower levels compared with that of GHtreated old-aged mice at 40 hours after PHx (Fig. 1A).
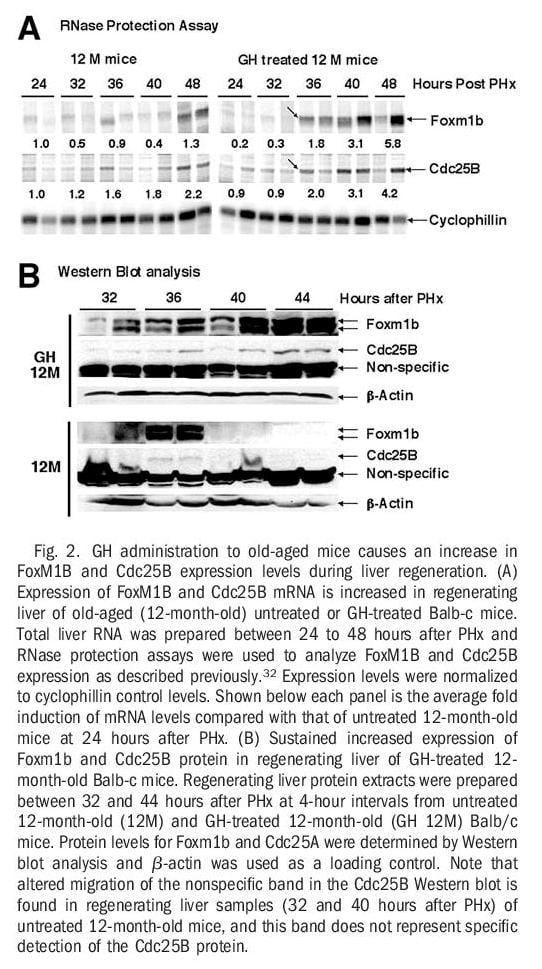
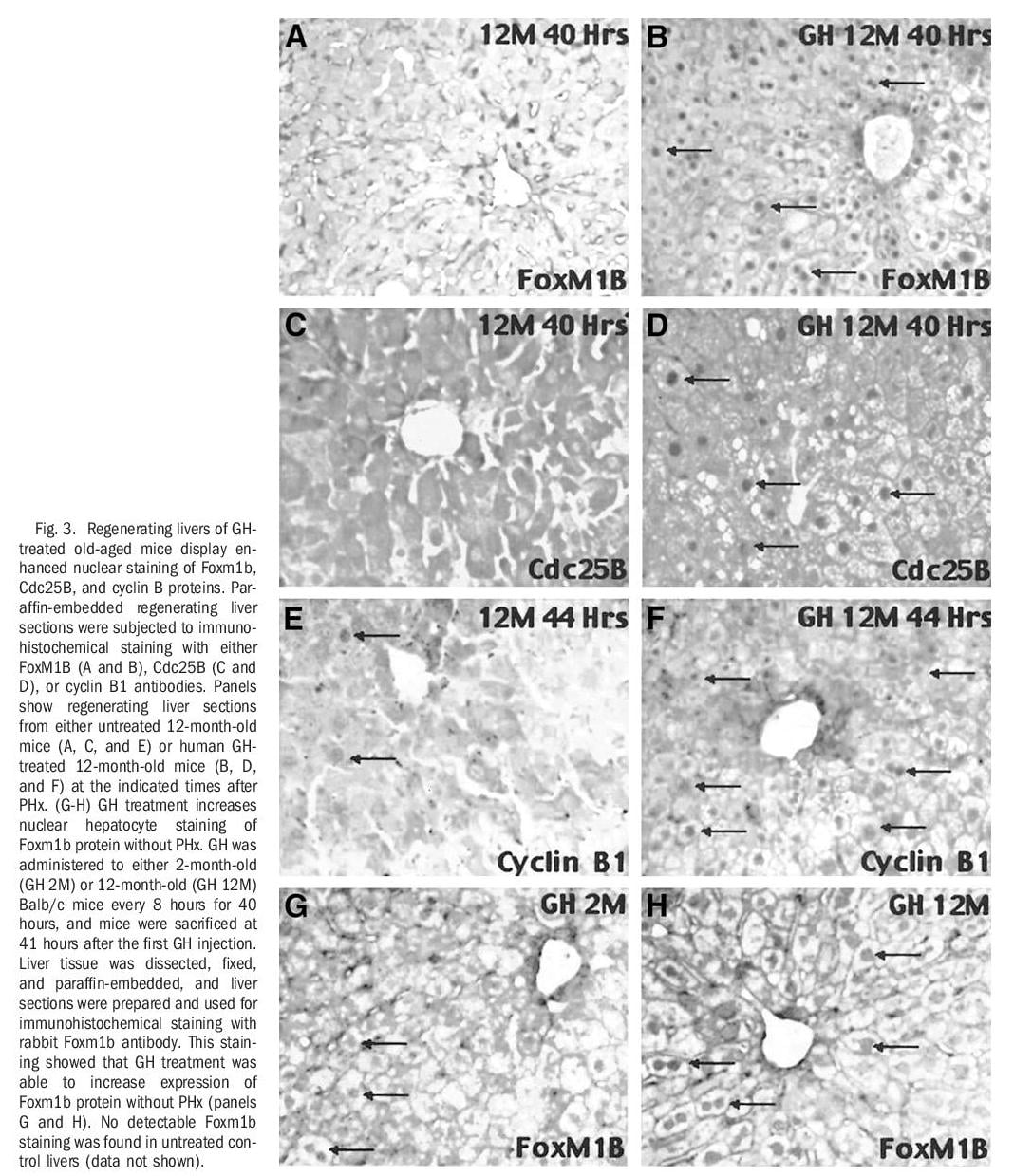
Human GH Treatment of Old-Aged Mice Stimulates Regenerating Hepatocyte Expression of Foxm1b, Cdc25B, and Cyclin B1. RNase protection assays revealed that GH treatment of old-aged mice increased expression of Foxm1b and its target gene Cdc25B at 36 hours after PHx and that these increased levels were sustained throughout the period of hepatocyte proliferation (Fig 2A). Western blot analysis with Foxm1b and Cdc25B antibodies showed that GH administration to old-aged mice caused a more substantial increase in hepatic protein levels of Foxm1b and Cdc25B between 40 and 44 hours after PHx compared with similar timepoints in untreated old-aged regenerating liver (Fig. 2B). Interestingly, untreated old-aged mice exhibited a transient increase in hepatic expression of Foxm1b and Cdc25B proteins at 36 hours after PHx, but these elevated protein levels were not sustained at the later timepoints after surgery (Fig. 2B). Furthermore, regenerating hepatocytes of GH-treated old-aged mice displayed a substantial increase in the nuclear staining of Foxm1b and Cdc25B proteins (Fig. 3B and D). This is compared with undetectable hepatocyte nuclear levels of these proteins in untreated old-aged mice at 40 hours after PHx (Fig. 3A and C). In addition, more substantial nuclear staining of cyclin B1 was observed in regenerating hepatocytes of GH-treated old-aged mice compared with untreated old-aged mice (Fig. 3E-F). Consistent with the ability of GH to induce hepatocyte DNA replication without PHx (Fig. 1E), increased hepatocyte nuclear staining of Foxm1b protein was found in either young or old-aged mice treated with GH alone (Fig. 2G-H). Hepatocyte nuclear staining of Foxm1b protein was undetectable in untreated mice (data not shown). These results suggested that GH administration is sufficient to induce Foxm1b protein expression in absence of liver injury. Taken together, these studies show that GH treatment of old-aged mice increased regenerating hepatocyte proliferation, expression, and nuclear localization of Foxm1b, Cdc25B, and cyclin B1 proteins.
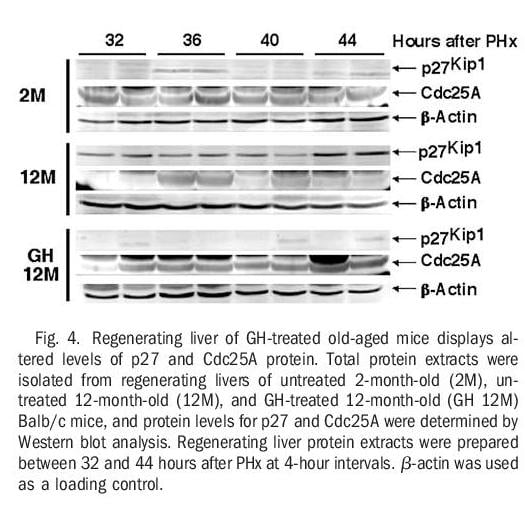
Regenerating Livers From GH-Treated Old-Aged Mice Displayed Diminished Levels of Cdk Inhibitor p27 and Increased Expression of Cdc25A Protein. Activation of S-phase promoting cyclin E-Cdk2 and cyclin A-Cdk2 complexes is mediated by Cdc25A dephosphorylation of specific Cdk2 protein residues and is inhibited by Cdk inhibitors, including p27.1-3,5,9,10 Our previous liver regeneration studies with old-aged Balb/c mice showed that increased regenerating hepatocyte expression of FoxM1B alone caused reduced levels of the Cdk inhibitor p27 protein.34 We next examined whether human GH treatment of old-aged mice could influence expression of both Cdc25A and p27 protein levels in regenerating liver extracts using Western blot analysis with commercially available p27 or Cdc25A antibodies (Fig. 4).
Young regenerating liver displayed only a transient increase in p27 protein levels at 36 hours after PHx and exhibited sustained expression of Cdc25A phosphatase protein throughout the period of hepatocyte proliferation (Fig. 4). Regenerating liver extracts from untreated oldaged mice exhibited a sustained increase in p27 protein levels and were unable to maintain high levels of Cdc25A protein beyond 36 hours after PHx (Fig. 4). In contrast, regenerating liver extracts from GH-treated old-aged mice exhibited a significant decrease in p27 protein levels and sustained expression of the Cdc25A phosphatase (Fig. 4). We have shown previously that both of these changes can be attributed to restoring Foxm1b levels in old-aged regenerating liver.33,34 Taken together, these studies show that GH treatment of old-aged mice was capable of increasing regenerating hepatocyte DNA replication by altering expression of proteins that stimulate Cdk2 activity, possibly through restoration of Foxm1b levels.
GH Treatment of Alb-Cre Foxm1b -/- Mice Failed to Restore Regenerating Hepatocyte Proliferation Resulting From Foxm1b Deficiency. Administration of GH has been shown to stimulate young hepatocyte proliferation after PHx.39 To genetically establish that GH required Foxm1b to increase regenerating hepatocyte proliferation, we determined whether GH treatment of Alb-Cre Foxm1b -/- mice could restore regenerating hepatocyte proliferation caused by Foxm1b deficiency. We measured hepatocyte DNA replication and mitosis in either untreated or GH-treated young AlbCre Foxm1b -/- mice at 40, 44, and 48 hours after PHx and compared regenerating hepatocyte proliferation with that of age-matched regenerating Foxm1b fl/fl liver (control). These liver regeneration studies showed that GH treatment of Alb-Cre Foxm1b -/- mice could not overcome the significant reduction in hepatocyte DNA replication or mitosis, and that hepatocyte proliferation levels were similar to those found in untreated regenerating AlbCre Foxm1b -/- livers (Fig. 5A and B). Consistent with the essential role of Foxm1b in transcriptional regulation of the Cdc25B phosphatase gene, administering human GH failed to increase expression of Cdc25B in Foxm1bdeficient regenerating livers (Fig. 5C). Our previous studies showed that regenerating Alb-Cre Foxm1b -/- hepatocytes displayed sustained increase in nuclear staining of Cdk inhibitor p21Cip1 (p21) protein (Fig. 5G-I), whereas regenerating Foxm1b fl/fl hepatocytes exhibited only a transient increase in p21 nuclear staining (Fig. 5D-F). Consistent with the role of Foxm1b in regulating p21 protein expression, GH treatment of Alb-Cre Foxm1b -/- mice was unable to overcome the persistent increase in regenerating hepatocyte nuclear staining of p21 protein (Fig. 5J-L). Taken together, these liver regeneration studies indicate that the presence of Foxm1b is essential for GH to stimulate regenerating hepatocyte proliferation.
Discussion
Reduced cellular proliferation during aging causes decreased tissue regeneration leading to diminished muscle and bone mass, defective tissue repair and thinning of the skin, which is associated with a progressive decline in GH secretion35-37 and FoxM1B expression.33,38 In a clinical study, GH therapy of older men was shown to reverse these decreases in muscle and bone mass and in skin thickness,37 but the transcription factors that are required for GH-mediated stimulation in proliferation remain uncharacterized. Previous liver regeneration studies showed that increased hepatocyte expression of FoxM1B alone in old-aged (12-month-old) mice was sufficient to stimulate regenerating hepatocyte DNA replication and mitosis to levels found in young (2-month-old) regenerating mouse liver.33,34 In this study, we showed that administration of human GH to old-aged Balb/c mice dramatically increased regenerating hepatocyte proliferation and expression of Foxm1b (Figs. 1, 2, 3B, and 6). This GH-mediated stimulation of regenerating hepatocyte proliferation in old-aged mice was associated with increased expression of Cdc25A and Cdc25B phosphatases and cyclin B1 protein with significant reduction of p27 protein levels (Figs. 3D, 4, and 6), several of which are functions previously attributed to restoring Foxm1b levels.33,34 These studies indicated that GH-mediated increase in FoxM1B levels stimulated cell cycle progression at both the S-G1 and G2-M phase transitions during liver regeneration. Furthermore, injection of GH alone was sufficient to stimulate hepatocyte proliferation and expression of Foxm1b protein, but induced lower levels of hepatocyte DNA replication than found in GH-treated mice after PHx. We provided strong genetic evidence that the presence of Foxm1b is essential for GH to stimulate regenerating hepatocyte proliferation, because GH treatment of young Alb-Cre Foxm1b -/- mice failed to restore regenerating hepatocyte DNA replication and mitosis resulting from Foxm1b deficiency.
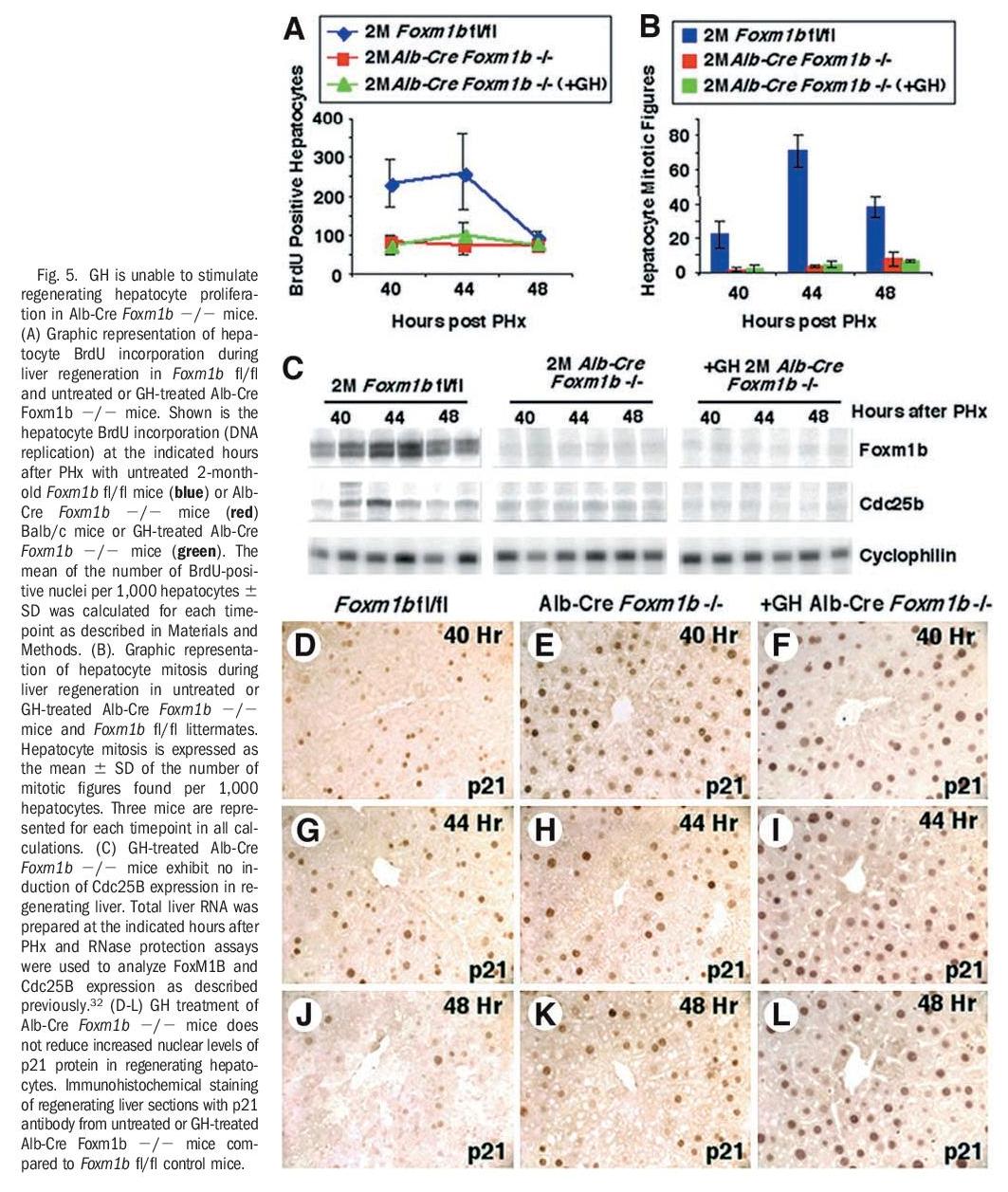
Our studies showed for the first time that increased levels of the Foxm1b transcription factor is essential for GH stimulation of regenerating hepatocyte proliferation in old-aged mice (Figs. 5 and 6).
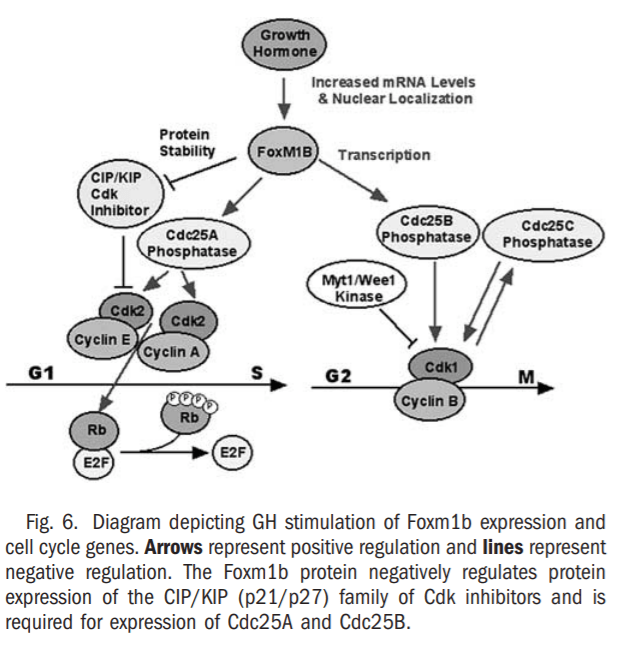
Binding of the GH receptor with ligand stimulates a number of cellular proliferation pathways that include the tyrosine Jak2 (Janus kinase 2)-Stat5A/B (signal transducers and activators of transcription), PI3K/Akt, Ras-mitogen activating protein kinase, and protein kinase C pathways.42 Our liver regeneration study suggests that hepatocytes from old-aged mice are still responsive to GH signaling, because increasing serum GH levels in old-aged mice was sufficient to restore regenerating hepatocyte proliferation. This result suggests that diminished proliferation during aging is not caused by a cell autonomous problem, but rather diminished secretion of GH, which is required in the serum to maintain normal cellular proliferation in response to injury. The use of Alb-Cre to mediate hepatocyte-specific deletion of the Foxm1b fl/fl targeted allele showed that the presence of Foxm1b is essential for GH stimulation of regenerating hepatocyte proliferation. GH-mediated activation of cell cycle signaling such as the Ras-MAP kinase and PI3K-Pdk1-Akt kinase pathways is likely responsible for increased FoxM1B expression in regenerating livers of old-aged mice. More recent studies have shown that FoxM1B transcriptional activity requires both Cdk-cyclin phosphorylation-dependent recruitment of p300/CBP and binding of activated Cdk-cyclin complexes to the activation domain (M. Major and R. H. Costa, manuscript submitted). Because activation of the Cdk-cyclin complexes requires both RasMAP kinase and PI3K-Pdk1-Akt kinase pathways, it is also likely that GH stimulation of these cell cycle signaling pathways mediates increased FoxM1b transcriptional activity. Our studies also suggest that GH administration may be an effective method to enhance hepatocyte proliferation in regenerating liver after either living donor or cadaver liver transplants. Moreover, our results indicate that GH administration is an effective method to enhance cellular proliferation in the older persons by increasing expression of Foxm1b, a transcription factor that is expressed in all proliferating cells.24-27
Although FoxM1B expression is induced greater than 40-fold during cellular proliferation,33,34 its promoter activity is increased only 4-fold in response to serum stimulation,43 suggesting that proliferative signaling also stimulates Foxm1b levels through increased messenger RNA (mRNA) stability. Consistent with this finding, deletion of the terminal 800 nucleotides from the 3’ end of the human FoxM1B complementary DNA resulted in stabilization of the FoxM1B transgene mRNA in quiescent mouse hepatocytes.16 We speculate that GH treatment of old-aged mice increases regenerating liver expression of Foxm1b, in part, through stabilization of Foxm1b mRNA. Furthermore, we found that the FoxM1B transgene protein was cytoplasmic in quiescent hepatocytes and proliferative signaling was required to induce hepatocyte nuclear translocation of the FoxM1B transgene protein after PHx.16 Although untreated oldaged regenerating liver expressed low levels of Foxm1b mRNA, nuclear staining of the Foxm1b protein was undetectable in these regenerating hepatocytes. In contrast, GH treatment of old-aged mice up-regulated Foxm1b mRNA and protein levels and also stimulated regenerating hepatocyte nuclear levels of Foxm1b protein, suggesting that GH signaling increased nuclear localization of the Foxm1b protein.
Stimulation of Cdk1 activity at the end of S-phase requires complex formation with cyclin B proteins and Cdc25B-mediated dephosphorylation of Cdk1 protein on Tyr15 and Thr14 residues.1-3, 44 Furthermore, microinjection of the Cdc25B protein, but not the Cdc25C protein, in tissue culture cells can drive replicating cells into mitosis.45 These and other studies suggest that Cdc25B is required to stimulate Cdk1 activity for progression from G2 phase into mitosis, whereas Cdc25C phosphatase is believed to function to maintain Cdk1 activity because activation of Cdc25C phosphatase requires Cdk1/cyclin B phosphorylation at mitosis.46,47 Our liver regeneration studies showed that GH treatment of old-aged mice was sufficient to increase expression and nuclear localization of Cdc25B phosphatase and cyclin B1 proteins in regenerating hepatocytes. Previous studies showed that regenerating Alb-Cre Foxm1b -/- livers display undetectable expression of Cdc25B and cotransfection assays show that FoxM1B stimulates the Cdc25B promoter.17 In our present study, we showed that GH treatment of Foxm1b -/- regenerating liver failed to stimulate expression of Cdc25B, suggesting that without the Foxm1b protein, GH itself is incapable of transcriptionally activating expression of the M-phase promoting Cdc25B gene.
Previously, it was shown that diminished regenerating hepatocyte DNA replication in old-aged Balb/c mice was associated with increased hepatocyte levels of the Cdk inhibitor p27 protein and that increased expression of Foxm1b was sufficient to diminish p27 protein levels.34 One possible explanation for increased nuclear levels of p27 protein is that one of the Foxm1b target genes is involved in mediating p27 protein degradation. Our current liver regeneration studies showed that GH treatment of old-aged mice caused diminished hepatic levels of the Cdk inhibitor p27 protein, a finding consistent with increased Foxm1b expression.34 Previous liver regeneration studies showed that Alb-Cre Foxm1b -/- hepatocytes displayed increased nuclear levels of p21 protein,17 suggesting that Foxm1b is regulating genes that are involved in p21 protein degradation. Consistent with these findings, GH treatment of young Alb-Cre Foxm1b -/- mice did not reduce the aberrant increase in regenerating hepatocyte nuclear staining of p21 protein caused by Foxm1b deficiency (Figs. 5 and 6). Moreover, we showed that regenerating liver of untreated old-aged mice displayed reduction in Cdc25A protein expression. Accumulating evidence shows that Cdc25A and Cdc25B proteins possess identical substrate specificity, and in either Cdc25B -/- or Cdc25C -/- mice, Cdc25A is believed to substitute for Cdc25B/C function.48,49 It is possible that reduction of Cdc25A protein levels in old-aged regenerating hepatocytes may prevent the ability of Cdc25A to substitute for Cdc25B function. Indeed, we showed that GH treatment of old-aged mice caused increased expression of both Cdc25A and Cdc25B phosphatases in regenerating liver, which is required for inducing Cdk activity and hepatocyte proliferation.
In summary, we showed that administration of human GH to old-aged mice restored regenerating hepatocyte proliferation and Foxm1b expression to levels found in young regenerating liver. This stimulation of regenerating hepatocyte proliferation was associated with increased expression of Cdc25A, Cdc25B, and cyclin B1 proteins with significant reduction in protein levels of the Cdk inhibitor p27, all of which are essential for appropriate cell cycle progression. Finally, these liver regeneration studies with hepatocyte-specific deletion of Foxm1b fl/fl targeted allele provided strong genetic evidence demonstrating that Foxm1b is essential for GH to stimulate regenerating hepatocyte proliferation.
- Nilsson I, Hoffmann I. Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res 2000;4:107-114.
- Sebastian B, Kakizuka A, Hunter T. Cdc25M2 activation of cyclin-dependent kinases by dephosphorylation of threonine-14 and tyrosine-15. Proc Natl Acad SciUSA 1993;90:3521-3524.
- Trembley JH, Ebbert JO, Kren BT, Steer CJ. Differential regulation of cyclin B1 RNA and protein expression during hepatocyte growth in vivo. Cell Growth Differ 1996;7:903-916.
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad SciUSA 1996; 93:13742-13747.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13:1501-1512.
- Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclindependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 1999;19:2109- 2117.
- Timchenko NA, Wilde M, Kosai KI, Heydari A, Bilyeu TA, Finegold MJ, Mohamedali K, et al. Regenerating livers of old rats contain high levels of C/EBP that correlate with altered expression of cell cycle associated proteins. Nucleic Acids Res 1998;26:3293-3299.
- Zhu L, Harlow E, Dynlacht BD. p107 uses a p21CIP1-related domain to bind cyclin/cdk2 and regulate interactions with E2F. Genes Dev 1995;9: 1740-1752.
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 2000;14:2393-2409.
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 2001;21:4684- 4699.
- Borgne A, Meijer L. Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J Biol Chem 1996;271:27847-27854.
- Wells NJ, Watanabe N, Tokusumi T, Jiang W, Verdecia MA, Hunter T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J Cell Sci 1999;112:3361-3371.
- Zachariae W, Nasmyth K. Whose end is destruction: cell division and the anaphase promoting complex. Genes Dev 1999;13:2039-2058.
- Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev 2000;14:142-146.
- Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF3/fork head DNA recognition motif resembles histone H5. Nature 1993; 364:412-420.
- Ye H, Holterman A, Yoo KW, Franks RR, Costa RH. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol Cell Biol 1999; 19:8570-8580.
- Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad SciUSA 2002; 99:16881-16886. HEPATOLOGY, Vol. 38, No. 6, 2003 KRUPCZAK-HOLLIS ET AL. 1561
- Costa RH, Kalinichenko VV, Lim L. Transcription factors in mouse lung development and function. Am J Physiol Lung Cell Mol Physiol 2001; 280:L823-L838.
- Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol 2002;250:1-23.
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci 2002;27:352-360.
- Ma Y, Geerdes DW, Vogt PK. Oncogenic transformation by the FOX protein Qin requires DNA binding. Oncogene 2000;19:4815-4821.
- Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol 2001;11:1950-1957.
- Nakae J, Biggs WH 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 2002;32:245-253.
- Korver W, Roose J, Clevers H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res 1997;25:1715-1719.
- Luscher-Firzlaff JM, Westendorf JM, Zwicker J, Burkhardt H, Henriksson M, Muller R, Pirollet F, et al. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: enhancement of transformation and transactivation. Oncogene 1999;18: 5620-5630.
- Yao KM, Sha M, Lu Z, Wong GG. Molecular analysis of a novel winged helix protein, WIN. Expression pattern, DNA binding property, and alternative splicing within the DNA binding domain. J Biol Chem 1997; 272:19827-19836.
- Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, Roebuck KA, et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol 1997;17:1626-1641.
- Diehl AM. Cytokine regulation of liver injury and repair. Immunol Rev 2000;174:160-171.
- Fausto N. Liver regeneration. J Hepatol 2000;32:19-31.
- Michalopoulos GK, DeFrances MC. Liver regeneration. Science 1997; 276:60-66.
- Taub R, Greenbaum LE, Peng Y. Transcriptional regulatory signals define cytokine-dependent and -independent pathways in liver regeneration. Semin Liver Dis 1999;19:117-127.
- Wang X, Hung N-J, Costa RH. Earlier expression of the transcription factor HFH 11B (FOXM1B) diminishes induction of p21CIP1/WAF1 levels and accelerates mouse hepatocyte entry into S-phase following carbon tetrachloride liver injury. HEPATOLOGY 2001;33:1404-1414.
- Wang X, Quail E, Hung N-J, Tan Y, Ye H, Costa RH. Increased levels of Forkhead Box M1B transcription factor in transgenic mouse hepatocytes prevents age-related proliferation defects in regenerating liver. Proc Natl Acad SciUSA 2001;98:11468-11473.
- Wang X, Krupczak-Hollis K, Tan Y, Dennewitz MB, Adami GR, Costa RH. Increased hepatic Forkhead Box M1B (FoxM1B) levels in old-aged mice stimulated liver regeneration through diminished p27Kip1 protein levels and increased Cdc25B expression. J Biol Chem 2002;277:44310- 44316.
- Cartee GD, Bohn EE, Gibson BT, Farrar RP. Growth hormone supplementation increases skeletal muscle mass of old male Fischer 344/brown Norway rats. J Gerontol A Biol Sci Med Sci 1996;51:B214-B2149.
- Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. J Lab Clin Med 2001;137:231-243.
- Rudman D, Feller AG, Nagraj HS, Gergans GA, Lalitha PY, Goldberg AF, Schlenker RA, et al. Effects of human growth hormone in men over 60 years old. N Engl J Med 1990;323:1-6.
- Ly DH, Lockhart DJ, Lerner RA, Schultz PG. Mitotic misregulation and human aging. Science 2000;287:2486-2492.
- Asakawa K, Hizuka N, Takano K, Horikawa R, Sukegawa I, Toyoda C, Shizume K. Human growth hormone stimulates liver regeneration in rats. J Endocrinol Invest 1989;12:343-347.
- Rausa FM, Tan Y, Zhou H, Yoo K, Stolz DB, Watkins S, Franks RR, et al. Elevated levels of HNF-3 in mouse hepatocytes influence expression of genes involved in bile acid and glucose homeostasis. Mol Cell Biol 2000; 20:8264-8282.
- Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000;26:149-150.
- Herrington J, Smit LS, Schwartz J, Carter-Su C. The role of STAT proteins in growth hormone signaling. Oncogene 2000;19:2585-2597.
- Korver W, Roose J, Heinen K, Weghuis DO, de Bruijn D, van Kessel AG, Clevers H. The human Trident/HFH-11/FKHL16 gene: structure, localization, and promoter characterization. Genomics 1997;46:435-442.
- Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I. The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci 1998;111:2445 2453.
- Karlsson C, Katich S, Hagting A, Hoffmann I, Pines J. Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J Cell Biol 1999;146:573-584.
- Izumi T, Maller JL. Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell 1993;4:1337- 1350.
- Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G. Phosphorylation and activation of human cdc25-C by cdc2: cyclin B and its involvement in the self-amplification of MPF at mitosis. Embo J 1993;12:53-63.
- Lincoln AJ, Wickramasinghe D, Stein P, Schultz RM, Palko ME, De Miguel MP, Tessarollo L, et al. Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat Genet 2002;30:446- 449.
- Chen MS, Hurov J, White LS, Woodford-Thomas T, Piwnica-Worms H. Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol Cell Biol 2001;21:3853-3861.

{kind=link}
{kind=link}
{kind=link}