
Liver and Iron Metabolism – A Comprehensive Hypothesis for the Pathogenesis of Genetic Hemochromatosis

Lipid overloading during liver regeneration causes delayed hepatocyte DNA replication by increasing ER stress in mice with simple hepatic steatosis
20.09.2019
Liver Regeneration and Aging: A Current Perspective
20.09.2019
Authors:W. Stremmel, M. Karner, E. Manzhalii, W. Gilles, T. Herrmann, U. Merle.
Abstract
It is hypothesized that a homozygous C282Y mutation of the HFE gene prohibits the assembly of the transferrin-receptor 1 (TFR1) with the divalent metal transporter (DMT1) as the main iron update complex in hepatocytes membrane. Thus, the cellular influx of transferrin-bound iron from the endosomal compartment into the cytasol is compromised. As a consequence, transferrin saturation increases while concomitantly a cytosolic iron deficiency state develops. This in turn triggers the suppression of hepcidin synthesis in hepatocytes. Its impaired release into the bloodstream, causes the increased intestinal iron absorption of hemochromatosis. Excessively absorbed iron cannot be used by the erythron as a surplus for hemoglobin synthesis and is therefore trapped in ferritin complexes of RES macrophages. The ferritin is thereafter released into the bloodstream and taken up by hepatocytes for final disposal. In the lysosomal compartment ferritin is degraded to hemosiderin. Here, the release of excessive iron molecules may induce cellular injury via free radicals. The phenotypic expression of genetic hemochromatosis may depend on the activity of the erythron to use transferrin-bound-iron for heme synthesis. Therefore, a high erythron requirement for iron can utilize excess iron and may represent the rationale of phlebotomy therapy in this disease.
The Physiology of Iron Metabolism
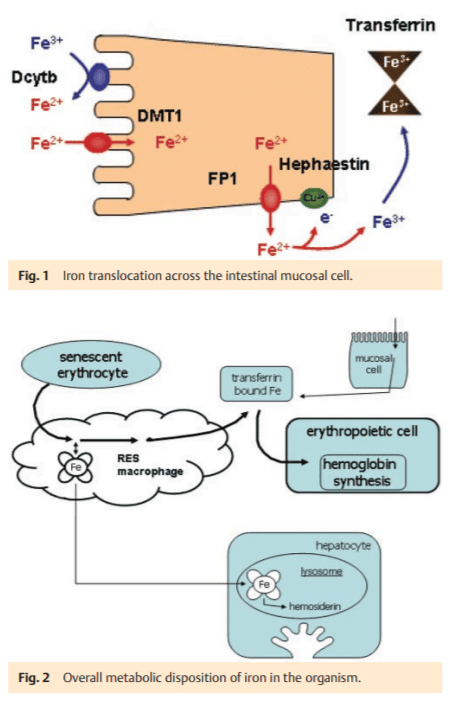
For the maintenance of a normal hemoglobin level, the body iron content has to allow for increased iron uptake in cases of iron deficiency anemia. Under physiological conditions, the loss of body iron is believed only to occur by passive cell desquamation, in particular by intestinal epithelial cells. It is primarily a constant loss of 1 mg per day. Thus, the control of body iron stores can only be mediated by a regulated absorption process. Intestinal uptake matches the demand of the body to maintain a balanced body iron content. The molecular arrangement of the cellular uptake process in duodenal mucosal cells involves several membrane-associated proteins, most of which are well-characterized (l » Fig. 1) [1].
At the luminal surface the iron reductase Dcyt b mediates the conversion of Fe3+ to Fe2+, which is the only form of iron that is able to pass through biological membranes. Translocation of Fe2+ across the apical plasma membrane is mediated by the divalent metal transporter 1 (DMT1, isoform 1) [2] which is non-selective for Fe2+ and also transports other divalent metals. The intracellular passage is incompletely understood and may involve cytosolic transport proteins, such as ferritin, or physiological chelators which bind iron in a non-toxic state. At the basolateral membrane, ferroportin 1 (FP1) transports Fe2+ across the membrane. Hephaestin (a ceruloplasmin-like protein), localized in the outer leaflet of the basolateral plasma membrane, oxidizes Fe2+ to Fe3+, thereby ensuring its binding to transferrin, the main transport protein of iron in blood. Transferrin delivers iron to the sites of its metabolism – mainly the erythropoietic system and the liver. Here, iron is taken up by the transferrin-receptor 1 endocytotic pathway. The release from this acidified endocytotic compartment into the cytosol is mediated by DMT1 (isoform 2) [2].
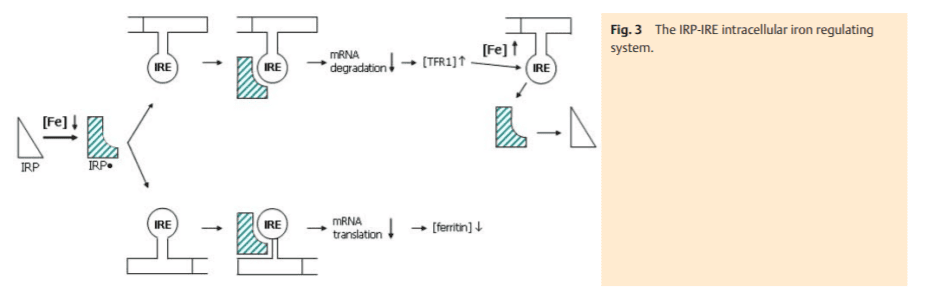
Iron is required in all cells in order to be incorporated into iron-containing proteins. Its presence is most important in the erythropoietic system where the delivery of iron corresponds directly to the demand for hemoglobin production [3]. Most of the iron required for the erythron is recycled from RES macrophages which recover the metal from senescent erythrocytes (Fig. 2) [3]. Within the RES cells, the hemoglobin-derived iron is translocated from phagosomes into cytosol by DMT1 (isoform 2) and, as described for the intestinal mucosa, is released from these cells by the FP1 pathway. At the outer leaflet of the plasma membrane Fe2+ is converted to Fe3+ by ceruloplasmin and again incorporated into transferrin for delivery to the sites of particular need, e. g., the erythropoietic cells. Excessive iron, originating by enhanced breakdown of erythrocytes in hemolysis or by increased absorption in hemochromatosis, is incorporated into ferritin by the RES macrophages. Ferritin is a Fe3+-clustering protein which by nature protects cells from any excess unbound iron and, thus, from free radical-induced injury (Fig. 2). It is assumed that iron can be released from ferritin in times of demand. In times of iron excess, however, iron-loaded ferritin is released into the circulation and rapidly cleared from circulation by yet incompletely understood mechanisms. This possibly involves a ferritin receptor-mediated uptake process in hepatocytes and subsequent lysosomal sequestration [4]. Whether iron can be released from hepatocytes into the bile for final disposal remains to be elucidated. The efficient hepatocellular clearance of ferritin is not affected by the hepatic iron content and this may be the reason why the liver contains a relatively high iron content which remains metabolically unused.
Regulation of Iron Metabolism
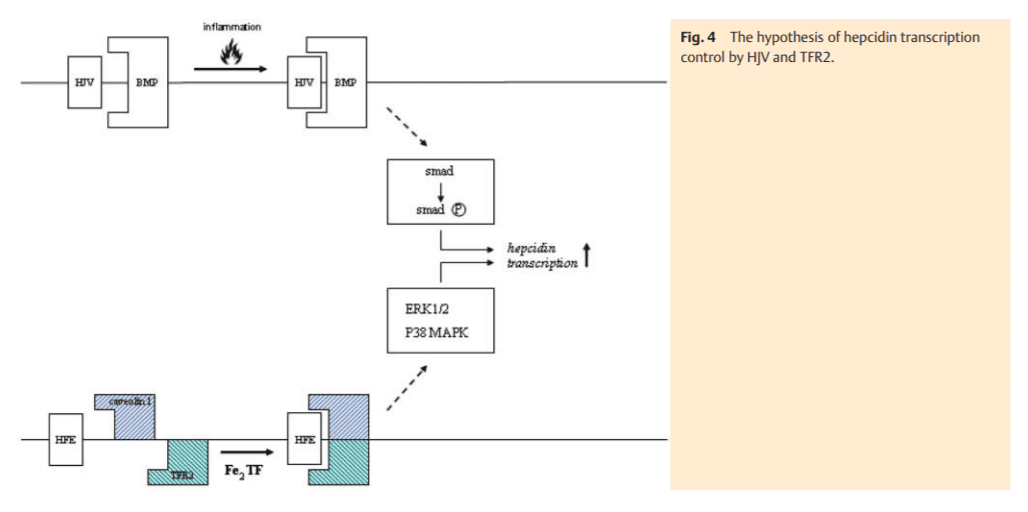
The uptake of transferrin-bound iron via transferrin-receptor 1 as well as its deposition in ferritin underlies a post-transcriptional control mechanism. It involves the intracellular iron-regulating proteins (IRP-1 and 2) which bind to a specific stemloop structure (iron responsive element-IRE) of the untranslated mRNA region (UTR) of the key iron proteins, i. e., TFR1 and ferritin (Fig. 3) [5]. IRP-1 is activated under conditions of cel lular iron demand. Binding to the 3’-IREs of the TFR1-mRNA inhibits its degradation by RNAses and consecutively stabilizes the mRNA. The resulting cellular TFR1 accumulation compensates for low cellular iron content. It is interesting that DMT1 (isoform 1), which resides in the plasma membrane of epithelial cells and is responsible for cellular uptake of non-transferrin bound iron, also contains a 3’-IRE [6]. In contrast, the isoform 2, which is particularly abundant in erythroid precursor cells and responsible for endosomal release of iron, has no IRE. The simultaneous IRP-1 binding to the 5’-IRE of the non-translated ferritin mRNA prohibits the transcription of this storage protein and subsequently any further cellular iron deposition – another aid in compensating cellular iron deficiency.
We believe that there is a protective mechanism that induces iron deficiency in inflammation. However, this mechanism is not fully understood. It is characterized on the one hand by low serum iron (low transferrin saturation with iron) and on the other hand by high serum ferritin levels. This implies that iron absorption is depressed and any excess of cellular iron is rapidly incorporated into the ferritin-“trap”. Inhibition of iron absorption is caused by a decrease of FP1 – the iron export pump present in intestinal mucosal cells. The FP1 is also highly abundant in RES cells. Thus, the common mechanism of FP1 blockade in inflammation leads to a reduced iron release from both of these cell types and consequently to an intracellular iron sequestration in the form of ferritin. From RES macrophages ferritin is released to serum and contributes to the ob served hyperferritinemia of inflammation. At the same time, transferrin saturation is low due to diminished iron absorption from duodenal mucosal cells. Anemia of chronic inflammation may be a result of this iron-depriving mechanism [7].
The major key player in this scenario is hepcidin, the iron regulatory “hormone”. Hepcidin is predominantly synthesized in hepatocytes. The expression of hepcidin is regulated by inflammatory signals, i. e., IL-6, leading to an increased secretion into bloodstream, whereas iron deficiency results in a suppressed hepcidin synthesis [8, 9]. It was a surprise that the hepcidin receptor was identified as ferroportin1 (FP1) – the above-mentioned cellular iron export pump – which is present in mucosal and RES cells as well as in placental cells and in minor quantities also in hepatocytes [10]. The binding of hepcidin to FP1 leads to the incorporation and degradation of FP1. The consequence is a lack of cellular release of free iron and its storage in the intracellular ferritin- “trap”. Thus, in the presence of high hepcidin levels iron, after its absorption into mucosal cells, is not translocated across the basolateral plasma membrane. This inhibition of iron absorption causes iron deficiency in the organism (resulting in a low transferrin saturation with iron).
The transcriptional control of hepcidin synthesis occurs via the upstream modulators hemojuvelin (HJV) and transferrin receptor 2 (TFR2) [11]. Both proteins reside in the plasma membrane and are linked to signalling pathways in order to control the transcription of hepcidin (Fig. 4). Hemojuvelin responds to inflammatory mediators by acting as bone morphogenetic protein (BMP)-co-receptor which leads to phosphorylation of the receptor-activated R-Smad transcription factor family. After binding Co-Smad, the R-Smad complex translocates to the nucleus where it then increases the transcription of hepcidin mRNA [12]. TFR2 is considered to be the sensor of the body’s iron status [13]. The latter is reflected by the amount of iron bound to transferrin (transferrin saturation). TFR2 binds (Fe)2-TF and conveys the iron status to a signal transduction effector-complex with downstream induction of hepcidin synthesis [14]. It is interesting to note that HFE binds to TFR2 in order to mediate the iron signal to the cell [14]. Moreover, a recent observation suggests that the membrane glycoprotein TFR2 is localized in caveolar microdomains interacting with CD81 and caveolin 1 [15]. This complex activates the “extracellular signal-regulated kinases” 1 and 2 (ERK 1 and 2) and p38 MAPK which in turn could control hepcidin synthesis (Fig. 4). This hypothesis can explain the dual regulation of hepcidin synthesis by iron via TFR2 and inflammatory signals via HJV.
Pathogenetic Concept of Genetic Hemochromatosis
In the pathological condition of genetic hemochromatosis, a moderate but constantly increased basal iron absorption leads to an increased body iron burden of 20 – 40 g (normal body iron content: 5 g) over a time period of 40 – 60 years.
This iron accumulation takes place in hepatocytes, whereas macrophages are generally spared. In most cases, this is caused by the homozygous C282Y mutation of the HFE gene. It is still not exactly clear how this mutation leads to an increased iron absorption. The normal HFE-protein binds in the ER to b2-microglobulin and then moves to the plasma membrane (Fig. 5), where it is integrated in the membrane and combines with transferrin-receptor 1 (TFR1). We hypothesize that DMT1 (isoform 2) also binds to this complex to mediate the cellular uptake of transferrin-bound iron. In fact, it has been shown in recent studies that HFE co-precipitates with DMT1, suggesting their close cellular interaction [16]. The C282Y mutation prevents the localization of HFE at the plasma membrane (Fig. 5). As a consequence, the assembly of the transferrin uptake complex HFETFR1-DMT1 is compromised. Iron-loaded transferrin may still bind to TFR1 and is incorporated by endocytosis. In the absence of HFE, however DMT1 may only randomly associate with the (Fe)2TF-TFR1 complex and therefore iron cannot be released from the coated pit endocytotic vesicle as efficiently. Thus the iron uptake into the cytosol is impaired. While this may not be as relevant in the erythropoietic system due to the high abundance of TFR1 and the isoform 2 DMT1 (in absence of HFE), it is of high significance in hepatocytes with a relatively low expression level of TFR1. Compromised iron release from endosomal vesicles due to an impaired TFR1-DMT1 association may be the reason why iron remains associated with transferrin. As a consequence, an iron deficiency state may result in hepatocytes (Fig. 6). This may, in turn, suppress hepcidin synthesis to compensate the perceived iron deficiency by the mechanisms described above.
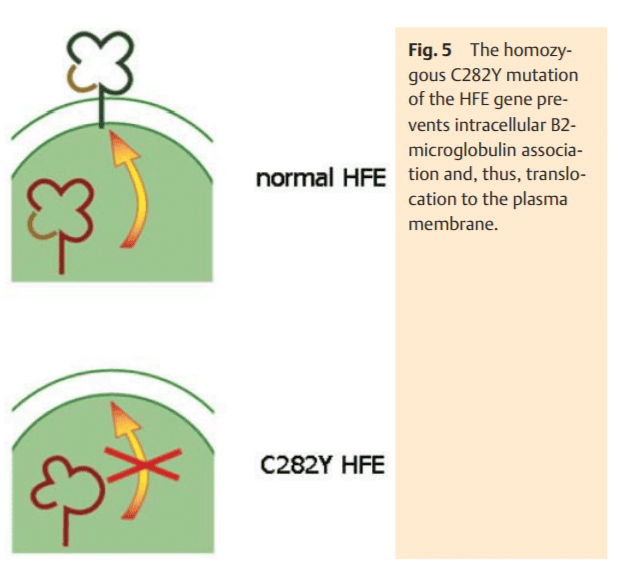
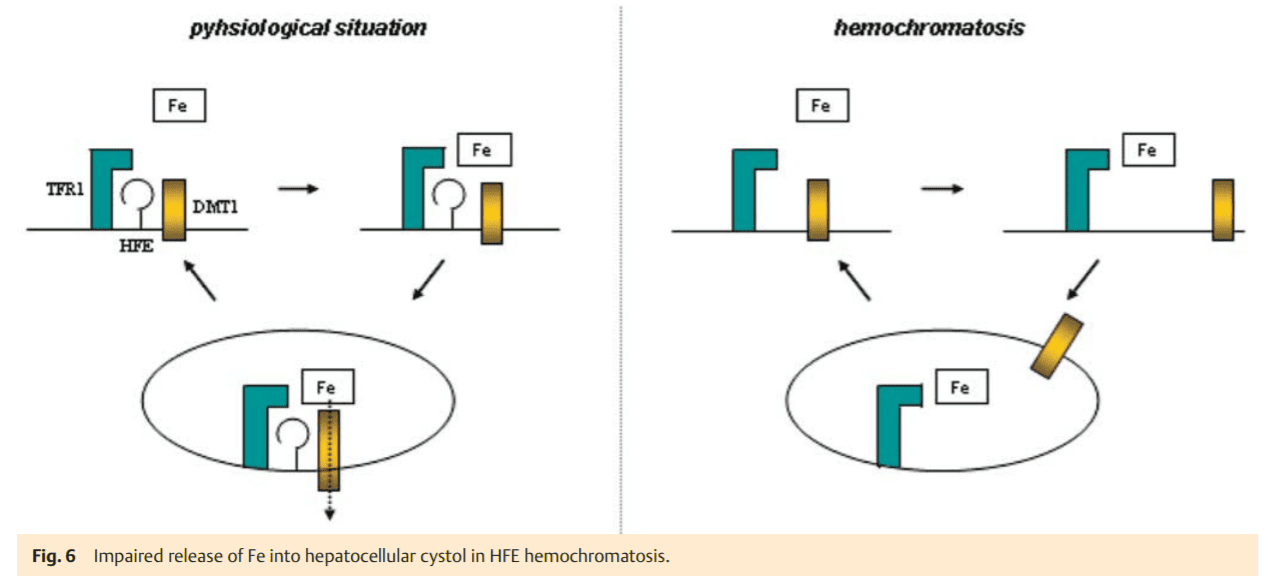
Over time, saturation of transferrin with iron increases which is a critical diagnostic feature in HFE-associated hemochromatosis. This is explained by the continuous cycling of transferrin bound iron through the endosomal compartment and its corresponding lack of transport in the cytosol mediated by DMT1. However, the feedback perception of highly saturated transferrin to TFR2 cannot be effective due to the lack of HFE (Fig. 4) [14]. Therefore, hepcidin transcription is further suppressed. As intestinal iron absorption is enhanced over a long period of time, an increase of body iron stores develops. Due to low hepcidin levels and a corresponding high FP1 activity, the RES cells are spared from iron loading in genetic hemochromatosis. The question arises, how the iron overload occurs in hepatocytes under these conditions where FP1 is also operative? Although this cannot be answered with certainty, it is obvious that excessively absorbed iron it not utilized by the erythron for surplus hemoglobin synthesis. Thus, it has to be incorporated into ferritin for storage, e. g., by RES cells. This ferritin with its inert and metabolically unusable iron is released into the bloodstream and taken up by hepatocytes for disposal. Since this iron is not registered as an excessive intracellular burden, the low hepatocellular iron signal caused by the HFE mutation persists. A repressed hepatic hepcidin excretion maintains the high iron absorption signal. Over the course of disease blood ferritin levels increase consistently, leading to hyperferritinemia – the other key diagnostic feature of genetic hemochromatosis. Hepatocytes accumulate excess ferritin which enters the lysosomal compartment, where it is tightly packed and may finally be degraded to hemosiderin. From there, free iron can be released and can induce free radical membrane injury which causes cellular damage. This as well as any other concomitant injury (e. g., by alcohol) induces an inflammatory response and hepcidin levels increase via the hemojuvelin pathway [12], counterbalancing enhanced iron absorption. At the same time the hepatocellular FP1 (the cellular iron export pump) is “closed” which deteriorates cellular free radical injury. The persistent hepatocellular damage causes cell death followed by liver regeneration – features which may lead to fibrosis and cirrhosis as well as promote hepatocarcinogenesis.
The current treatment of genetic HFE-associated hemochromatosis by phlebotomies leads to a decrease of the body iron burden. Thus, ferritin production for the deposition of excessive iron ceases and serum ferritin levels drop concomitantly. Iron is now more urgently required by the erythron for hemoglobin production. Indeed, it is the high demand for iron by the erythron which channels the absorbed transferrin-bound iron to hemoglobin synthesis and prohibits deposition into ferritin. The high iron demand of the erythron may also be the reason why menstruating women rarely develop severe iron overload. Moreover, the individual differences in iron requirement may explain the variable clinical penetrance of HFE-associated hemochromatosis. Genetically-susceptible individuals (homozygous C282Y mutations) with a low iron demand develop iron overload, whereas those with high demand do not.
Non-HFE-Related Hemochromatosis
Non-HFE-related hemochromatosis is exceptionally rare and is caused by mutations of the HJV, TFR2 or hepcidin (HAMP) genes [3]. The iron absorption rate is much higher compared to the HFE-associated hemochromatosis and results in more pronounced iron overload [17, 18]. Obviously, the suppression of the hepcidin level is most severe in cases of HAMP mutations. HJV and TFR2 gene mutations also induce very low hepcidin levels due to the fact that they are the two directly connected upstream regulators of hepcidin synthesis (Fig. 4). The clinical manifestation of these severe iron overload disorders occurs already in early adulthood (juvenile hemochromatosis) and in addition to the liver often involves the heart with a cardiomyopathy. The iron deposition in the B-cells of the pancreas is of a destructive character and often not reversible. It is responsible for the occurrence of an insulin-dependent diabetes. In addition to that, hyperinsulinemic diabetes can develop due to an impaired clearance of insulin by injured he patocytes in advanced stages of the disease. This, however, can be improved by phlebotomies. Iron deposition in gonadotropic cells of the pituitary gland results in hypogonadotrophic hypogonadism. This rather rare clinical feature is more common in the non-HFE hemochromatosis population. Clearly independent of the underlying mutation, the destructive arthropathy is a typical feature in HFE-related and non-related hemochromatosis and can even appear during therapeutic phlebotomy. The pathogenetic nature of this arthropathy which involves metacarpophangeal and weight-bearing joints remains unclear. It has been speculated that it might be caused by an iron-induced inhibition of pyrophosphorylases in synovial cells leading to deposition of pyrophosphate crystals in the joint cavity.
1 Sharma N, Butterworth J, Cooper BT et al. The emerging role of the liver in iron metabolism. Am J Gastroenterol 2005; 100: 201–206
2 Lam-Yuk-Tseung S, Gros P. Distinct targeting and recycling properties of two isoforms of the iron transporter DMT1 (NRAMP2, Slc11A2). Biochemistry 2006; 45: 2294–2301
3 Pietrangelo A. Hereditary hemochromatosis – a new look at an old disease. N Engl J Med 2004; 350: 2383–2397
4 Mack U, Powell LW, Halliday JW. A receptor for ferritin on hepatocytes. J Biol Chem 1983; 258: 4672–4675
5 Hentze MW, Caughman SW, Casey JL et al. A model for the structure and functions of iron-responsive elements. Gene 1988; 72: 201–208
6 Johnson DM, Yamaji S, Tennant J et al. Regulation of divalent metal transporter expression in human intestinal epithelial cells following exposure to non-haem iron. FEBS Lett 2005; 579: 1923–1929
7 Andrews NC. Anemia of inflammation. The cytokine-hepcidin link. J Clin Invest 2004; 113: 1251–1253
8 Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102: 783–788
9 Nemeth E, Rivera S, Gabayan V et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271–1276
10 Nemeth E, Tuttle MS, Powelson J et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306: 2090–2093
11 Pietrangelo A. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut 2006; 55: 564–568
12 Babitt JL, Huang FW, Wrighting DM et al. Bone morphogenetic protein signalling by hemojuvelin regulates hepcidin expression. Nature Genetics 2006; 38: 531–539
13 Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 2004; 104: 4287–4293
14 Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem 2006; in press (manuscript C 600197200)
15 Calzolari A, Raggi C, Deaglio S et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J Cell Science 2006; in press (online publication, 17 October 2006)
16 Arredondo M, Tapia V, Rojas A et al. Apical distribution of HFE-beta2- microglobulin is associated with inhibition of apical iron uptake in intestinal epithelial cells. Biometals 2006; 19: 379–388
17 Schilsky ML, Fink S. Inherited metabolic liver disease. Curr Opin Gastroenterol 2006; 22: 215–222
18 Pietrangelo A, Caleffi A, Henrion J et al. Juvenile hemochromatosis associated with pathogenetic mutations of adult hemochromatosis genes. Gastroenterology 2005; 128: 470–479

{kind=link}
{kind=link}
{kind=link}